
Webpages about Smoldyn and spatial simulation
- Brief description of Smoldyn
- Spatial simulation and simulators
- Comparison table of particle-based simulators
- Questions and answers about Smoldyn
- Advice on choosing simulation parameters
- Research project ideas
- Algorithm details
Simulator comparison
This table compares all particle-based simulators that I am aware of which have had releases in the past 3 years. It was accurate, to the best of my knowledge, as of November 2017.
Click on any row for details (sorry, this feature doesn't work in Internet Explorer).
Versions: MCell 3.4, Smoldyn 2.54, eGFRD 2.1, SpringSaLaD 2016 release, ReaDDy 2017 release.
Time steps
~1 μs
ns to ms
event-based
~10 ns
~0.1 ns to μs
MCell Version 3 introduced a scheduling-based system in which "the scheduler maintains a user-specified canonical time step ΔT during the simulation, but allows an internal Δt to vary from molecular species to molecular species" [1]. While not as sophisticated as GFRD, this scheduling-based system should nevertheless be more computationally efficient than can be achieved with standard fixed time steps. Most MCell simulation used a time step of about 1 μs (e.g. [2-4]).
Smoldyn uses fixed time steps [5]. It has been used with time steps ranging from 0.06 ns to 20 ms. It is most often used with time steps ranging from 10 μs to 1 ms for systems biology research and 1 ns to 1 μs for biophysics research. For advice on choosing time steps, see [6]. Smoldyn also allows the time step length to be changed mid-simulation, including with a dependency on current molecule count values.
eGFRD time steps are event-based, in each case stepping from the exact moment of one event to that of the next event [7-9], where events include molecule-molecule collisions, chemical reactions, and molecule-surface interactions (although surfaces are not implemented in the current software). Only the individual molecules that participated in the events are updated at each time step, with the result that the system state always represents information from many different times at once.
SpringSaLaD simulates bonds between different protein "sites" using springs. They can generally be simulated accurately when time steps are between 1 and 100 ns for biologically relevant diffusion coefficients [10]; 10 ns is recommended as a typical time step.
ReaDDy can use a wide range of time steps and, as usual, shorter steps yield better spatial resolution. However, ReaDDy was designed particularly for investigating the effects of particle interaction potentials, including the effects of space exclusion, molecular crowding, and aggregation. Investigating these phenomena requires that time steps are short enough that a molecule has multiple steps along a curved potential energy surface [11]. In one test, reported in the same paper, a good value was found to be 0.1 ns.
Molecules
points
points, spheres
spheres
multi-spheres
multi-spheres
MCell molecules have a fixed "interaction radius" that applies to all molecules in the simulation. However, non-reactive molecules do not interact with each other, making them effectively points.
Smoldyn molecules can be represented as simple points, so that non-reactive molecules simply pass through each other without any interaction [12]. This allows for fast simulations and is an appropriate representation whenever the modeled molecules occupy a small fraction of the local volume. Smoldyn also allows molecules to be represented as spheres, thus allowing for excluded volume interactions [13].
eGFRD molecules are always spheres with excluded volume.
SpringSaLaD molecules are collections of spheres, each of which is called a "site", that are connected with spring-like bonds [10]. It is a strictly netword-free simulator, so it is not possible to specify a reaction network directly. Instead, the user defines sites along with their binding interactions. This constraint makes physical sense but does require a somewhat different modeling approach than is used with other simulators (e.g. the Michaelis-Menten reaction, E+S ↔ ES → E+P, needs to be expanded to E+S ↔ ES → EP → E+P).
ReaDDy molecules are aggregates of spheres [11], connected through interaction potentials.
Dimensions
2,3
1,2,3
3
3
3
Low-dimensional (1D and 2D) simulations can be useful, for example, for investigating protein diffusion along a nucleic acid filament or interaction rates of integral membrane proteins.
MCell always stores 3D coordinates for molecules and surface tiles so all simulations are 3D in a sense. However, it's possible to confine the system to a 2D plane. MCell supports reactions between molecules that are surface-bound so it's possible to run simulations in which everything takes place on a surface.
Smoldyn simulations can be run in 1, 2, or 3 dimensions [5]. These are true low-dimensional simulations because Smoldyn only stores as many coordinates as the system dimensionality. As a result, Smoldyn simulates low-dimensional simulations much faster than 3D simulations. Smoldyn supports reactions, excluded volume, and other interactions between molecules that are in low dimensions.
eGFRD methods have been developed for 1D and 2D systems as well [14], but are not implemented in the current software.
System boundaries
ref.,abs.,per.,trans.
ref.,abs.,per.,trans.
periodic
reflect
per.,interact.
ref. = reflecting, abs. = absorbing, per. = periodic, trans. = transparent, interact. = interacting
System boundaries are called periodic if molecules that diffuse out of one side of the system then diffuse into the opposite side of the system. Periodic boundaries are useful for efficiently simulating large homogeneous systems without incurring unwanted effects from reflective system boundaries.
Transparent boundaries allow molecules to diffuse out of the system boundaries. Even outside the boundaries, these molecules are still tracked and can still react or interact with surfaces.
MCell supports two types of periodic boundaries. Traditional boundaries are the normal type in which molecules that diffuse off of one side of the simulation volume diffuse onto the other side. In the alternate mirrored boundaries, "the geometry is mirrored in adjacent boxes and molecules do not wrap around. When a molecule hits a boundary, it will continue diffusing in the mirrored adjacent boundary." (see here).
SpringSaLaD only supports a rectangular domain that is partitioned into intracellular space, extracellular space, and a membrane; it is surrounded by a reflecting boundary [10].
ReaDDy supports periodic and softly replusing boundaries.
Surfaces
triangle mesh
many primitives
-
1 flat surface
plane, sphere
MCell surfaces are represented as lists triangles, called either "elements" or "tiles," which can then be used to create quite complicated surface shapes (e.g. [4]). Triangle-shaped primitives are sufficient to define any shape of surface [15]. MCell does not support additional primitive shapes, although it allows the user to specify boxes in the configuration file for convenience, which it represents internally as triangles. Note that MCell only allows a single molecule per tile, which may promote the use of a finer surface meshing.
Smoldyn surfaces are represented as lists of geometry primitives called "panels" [5]. The panel shapes for 3D simulations can be any of: axis-aligned rectangle, triangle, sphere, cylinder, hemisphere, or disk. Fewer options are permitted for 1D and 2D. This range of panel shapes simplifies input file development and also speeds up simulations. As in MCell, arbitrarily complex shapes can be constructed using lists of triangles.
eGFRD does not support simulations with internal surfaces.
SpringSaLaD geometry is fixed to a rectangular domain with a single internal planar surface that represents a cell membrane and which separates intracellular and extracellular regions [10].
ReaDDy supports planar and spherical surfaces that can be defined within the simulation volume.
Surface molecules
≤1 molec., 2 states
unlimited, 4 states
-
unlimited, 3 states
-
MCell allows up to 1 molecule per surface element (triangular tile). When MCell simulates surface diffusion, these molecules have fixed point-like positions. However, when MCell simulates chemical reactions, it does not consider the exact positions of these molecules, but instead treats each molecule as though it is at a random position within its tile. These limitations, which are rarely important, can be minimized by constructing surfaces from very small tiles. MCell surface molecules may be oriented in either the "top-front" or "top-back" states to distinguish the two transmembrane orientations.
Smoldyn allows unlimited surface molecule density on any panel. Smoldyn supports four surface molecule "states" [5]. They were originally designed so that that the "up" and "down" would represent two orientations of trans-membrane proteins and the "front" and "back" states would represent molecules that are adsorbed to the front or back of a surface. In practice, all four states are all essentially equivalent.
SpringSaLaD allows molecules to be bound to the membrane. Their density is limited by their excluded volume but not in other ways. SpringSaLaD designates molecules as being intracellular, extracellular, or membrane-bound [10]. Membrane-bound molecules are attached to anchors that are in the membrane but do not have additional orientation information. However, they can be exposed to one side exclusively, the other side, or both sides, thus leading to 3 effective states.
ReaDDy does not support molecules in distinct surface-bound states. However, molecules can interact with surfaces through interaction potentials, and hence stick through these forces.
Excluded volume
-
excellent
exact
good
excellent
Smoldyn typically does not simulate excluded volume interactions, which makes simulations much faster. However, it can simulate them, and does so quite accurately if so. Smoldyn accounts for excluded volume by checking for any molecules that overlap each other at the end of a time step and then moving them to correct the overlap. It moves them using either an "overlap" or "reflection" method [13]. In the former case, the molecules are separated by a distance that equals the original overlap distance. In the latter case, the straight-line trajectories of the two molecules are computed as they collided, and the trajectories are then reflected off of each other. Both algorithms are exact in the limit of short time steps. They enabled very close agreement with exact theory for a simulation of a hard sphere fluid [13].
eGFRD simulates excluded volume interactions exactly. However, they can be quite slow [16]. I have not seen a validation of excluded volume simulation. If eGFRD is started with too much of the simulation volume occupied by randomly placed molecules, it can search indefinitely for good placement positions.
SpringSaLaD accounts for excluded volume using a hard sphere type of model. However, simulation of a hard sphere fluid with SpringSaLaD showed that the resulting radial distribution function showed more structure than it should, which was eventually traced to the fact that SpringSaLaD places molecular collision products adjacent to each other rather than at a finite separation (personal communication with Paul Michalski, October 2016). This effect should shrink as time steps are reduced to zero but, in practice, I found substantial errors with even very short time steps [13].
ReaDDy was designed in part for simulating excluded volume interactions (although accounting for excluded volume is not required). Figure 6 of [11] shows excellent agreement between ReaDDy and a molecular dynamics simulation. Note that ReaDDy is designed for an interaction potential between molecules; this enables more realistic excluded volume interactions than are possible with hard-sphere assumptions, but this also may mean that ReaDDy is not optimized for hard sphere interactions.
Multimers
states only
rule-based model
-
explicit
explicit
MCell does not support rule-based modeling but does support molecules that have internal states, such as being phosphorylated. These states are represented with molecule "slots" [17].
Smoldyn supports two types of rule-based modeling [13]. Rules can be expressed in the BNGL language, which Smoldyn then sends to the BioNetGen software for expansion into reactions. This is only available with the generate-first approach. Rules can also be expressed in a wildcard method, where these wildcards are similar to those used in computer operating systems. Several sample models showed that the wildcard method is remarkably convenient and powerful [18].
SpringSaLaD represents multimers explicitly with each of the separate components, represented as a sphere, and their bindings. It calls the components "sites"; each site can be one of several user-defined "types". In addition, the sites can have multiple states.
ReaDDy also represents multimers explicitly, with a sphere for each of the separate components. These spheres are bound together by potential energy interactions.
Allostery
-
yes
-
yes
-
Smoldyn supports allosteric reactions, which are a special type of bimolecular reactions. Here, a chemical reaction occurs with first-order kinetics if a molecule of one species is sufficiently close to a molecule of a second (or the same) species. The reaction products are typically placed at the reactant locations. Allosteric reactions are useful for simulating the spread of activity across a tightly coupled cluster of receptor proteins [19], microtubule growth, or many other types of dynamics.
SpringSaLaD supports reactions in which the state of one particle within a multimeric complex affects the reactions of other particles within the same complex (from the Supplementary Information of [10]).
Reaction accuracy
very good
excellent
exact*
excellent
excellent
* The eGFRD algorithm is exact but the software doesn't simulate reaction rates correctly at present.
MCell simulates bimolecular reactions with the assumption that molecules travel in straight lines during time steps [1] and interact with reactants whose center is within a molecular diameter of this trajectory; these interactions can lead to reactions, based on a pre-computed reaction probability. This algorithm does not match reality because solution-phase molecules do not actually travel in straight lines, but this has a negligible effect on simulation accuracy. However, the reaction probabilities are calculated with the assumption that reactants are always well-mixed, which leads MCell to simulate bimolecular reaction rates too slowly [5]. The reason is that reactants that are close to each other generally react fairly quickly, leaving behind those that are more widely separated. Thus, at steady-state, the local region around each reactant is depleted of the molecules of the other species, which violates the well-mixed assumption. As a result, MCell is generally fairly accurate when simulations use long time steps, slow reaction rate constants, and large interaction radii but becomes increasingly in error as these parameters are changed in the other direction. I described the errors in ref. [5], but newer results have shown that the errors can be largely mitigated by using interaction radii that are much larger than k/(4πD) where k is the reaction rate constant and D is the sum of the reactant diffusion coefficients. Test file: MCellBireact.txt (change the suffix to mdl to run the simulation).
Smoldyn's default bimolecular algorithm simulates a bimolecular reaction whenever two reactants end up within a pre-determined "binding radius" of each other at the end of a time step, and not otherwise. The rational for this algorithm is that it is extremely fast and that the simulation only needs to be accurate at the ends of the time steps, so that any dynamics that arise during the time steps are not meaningful. Smoldyn computes the binding radius for a steady-state reactant distribution, rather than a well-mixed one, which yields reaction rates that are within about 2.5% of the exact results for any length time step [12]. Several tests have further confirmed this accuracy [5]. Smoldyn's reaction dynamics generally agree well with those of the Collins and Kimball model, and not the Smoluchowski model for which Smoldyn is named (and which the reaction dynamics approach in the limit of short time steps). Despite common misconceptions (e.g. [14, 20, 21]), this algorithm does not lead to binding radii that are much smaller than the molecules themselves but that are instead reasonably close to molecular radii [5]. Note that Smoldyn supports the Doi algorithm, in which there is a specific reaction probability for overlapping reactants. However, reaction rates have not been calibrated yet for this algorithm. Test file: SmoldynBireact.txt.
eGFRD computes reaction rates and reaction dynamics exactly, in principle [9]. Exact simulation means that the dynamics exactly match those of the Collins and Kimball reaction model [22]. If the user enters a molecule radius that is too small (meaning that it is less than k/(4*pi*D), where k is the reaction rate constant and D is the mutual diffusion coefficient), then it is impossible for the eGFRD algorithm to yield a fast enough reaction rate so it simulates it with the slower diffusion-limited reaction rate. More problematic is that my tests of the eGFRD simulator showed that the actual simulated reaction rates were only about 66% of the theoretical mass-action reaction rate, which presumably arises from a bug in the software. Also, I was unable to test the fast reaction rate with eGFRD because this required molecules with at least 2 nm radii, and eGFRD wasn't able to place molecules of this size and the requested density in the simulation volume in a reasonable amount of time. Test file for medium reaction rate: eGFRDBireact.txt (change the suffix to gfrd to run the simulation).
SpringSaLaD simulates second order reactions using an improvement on Erban and Chapmans λ-ρ theory [20]. The original theory is very accurate for molecules that do not have excluded volume but is in error for molecules with excluded volume. SpringSaLaD uses an algorithm that corrects for this issue [10].
ReaDDy simulates second order reactions using Erban and Chapman's λ-ρ theory. I tested its accuracy using the same A + B -> C model with which I tested the other simulators and found excellent agreement with mass action theory. In this simulation, ReaDDy did not account for molecule excluded volume. Test file: ReaDDyBireact.txt (change the suffix to py to run the simulation).
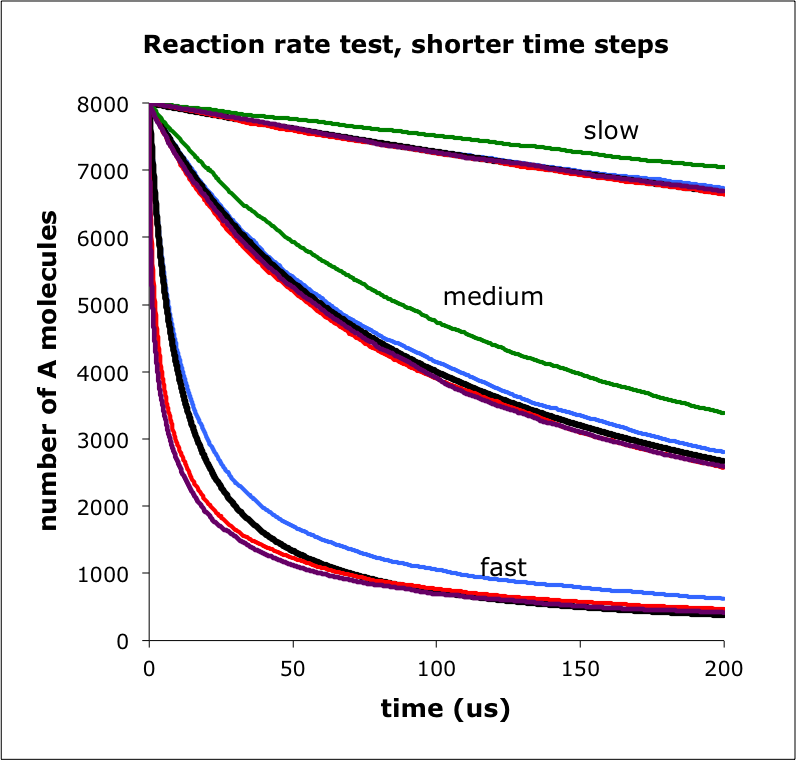
The figure shows the number of A molecules over time for a simulation of A+B -> C. I ran the simulation using 3 rate constants, labeled slow, medium, and fast. For each rate constant, the lines are: black is mass action, blue is MCell, red is Smoldyn, green is eGFRD, and purple is ReaDDy. Focusing on the fast reactions, Smoldyn and ReaDDy are faster than mass action at short times because the molecules are randomly placed at the simulation start; this leads to a faster than mass action reaction rate, as the molecules approach the steady-state distribution, for which mass action then applies.
Dissociation products
stochastic
fixed separation
adjacent
adjacent
adjacent
If dissociation reactions are reversible, this means that the individual product molecules of the dissociation can undergo an association reaction with each other, which is called a geminate recombination. Geminate recombinations occur with some finite probability with real molecules, so their probability needs to be controlled in simulations as well.
MCell accounts for geminate recombinations by allowing simulations to run with a "microscopic reversibility" feature [1]. When this is activated, dissociation products are initially separated by a random distance that is chosen from a probability density that is calculated to match that of the reactants before they combined to form a complex. This is done so the simulator obeys detailed balance.
Smoldyn accounts for the effect of geminate recombinations on overall reaction rates by initially separating dissociation products by a fixed "unbinding radius" [12]. This produces accurate reaction rates and equilibrium concentrations. It leads to a slight violation of microscopic reversibility (detailed balance) for the region between the binding and unbinding radii, but detailed balance is still obeyed for shorter and larger distances. Because interaction potentials between molecules do not actually obey the Collins and Kimball model on small distance scales anyhow, the failure to obey detailed balance in this region is irrelevant for almost all applications [23].
eGFRD does not separate dissociation products, but places them adjacent to each other [7]. This is correct because the simulation time steps exactly up to the dissociation event, meaning that the products have not had time to diffuse away from each other afterwards. eGFRD controls the probability of geminate recombination by using the Collins and Kimball reaction model [22], in which there is a finite intrinsic reaction rate, which is the reaction rate of particles that are in contact with each other [24].
SpringSaLaD does not move dissociation products but simply removes the bond between them. The bimolecular reaction probability has an explicit dependence on the bond length to automatically account for geminate recombination [10].
ReaDDy places dissociation products adjacent to each other. This does not result in certain geminate recombinations because ReaDDy performs bimolecular reactions with an intrinsic rate constant, much like GFRD.
Molec.-surf. interactions
good
excellent
-
to sites only
potentials
MCell detects molecules that diffused across a surface during the previous time step and then adsorbs it to the surface according to a pre-computed probability. MCell calculates the probability by treating adsorption like a reaction between a volume molecule and a surface-bound molecule. As a result, the "rate constant" for adsorption in MCell has units of M-1s-1, which does not match the length/time units that ought to be used for adsorption coefficients. However, if MCell's adsorption reaction rate constant is divided by the area for each surface tile, then the MCell probability equation [1] (eq. 4.7) matches the analogous probability that was derived by Erban and Chapman ([25] eq. 10). This derivation assumes a well-mixed molecular distribution, which is not quite correct because it ignores the effect of adsorption on the distribution.
Smoldyn also detects molecules that diffused across a surface during the previous time step and then adsorbs it to the surface according to a pre-computed probability. It achieves essentially exact adsorption rates by calculating the adsorption probability for a steady-state molecular distribution [26]. The Smoldyn algorithm also explicitly accounts for the differences between irreversible and reversible adsorption. Smoldyn desorbs molecules with an initial separation between the desorbed molecule and the surface that is randomly chosen from the appropriate probability density for reversible or irreversible desorption [26].
SpringSaLaD does not simulate adsorption, desorption, or membrane transmission. However, if an anchor is already placed on a surface, then additional molecules can bind to or unbind from this anchor.
ReaDDy interactions between molecules and surfaces are exclusively through potential energy interactions. These interactions enable adsorption, desorption, and transmission. The user cannot enter surface interaction rates as rate constants, but instead enters them using potential energy interaction parameters.
Long-range interactions
-
-
-
-
yes
Long-range interactions are forces between molecules that extend beyond the distances at which they physically contact each other. For example, electrostatic forces and van der Waals forces are long range interactions. The strengths of these forces falls off rapidly in biological systems due to Debye screening by ions, with a typical screening distance of about 1 nm. Thus, electrostatic interactions can usually be ignored if distance scales of interest are all substantially greater than 1 nm.
SpringSaLaD does not account for long-range interactions between unbound molecules. However, if molecules are bound together with a bond, then they interact through a spring potential.
ReaDDy works at a higher level of detail than the other simulators in part because it accounts for interaction potentials between molecules, and between the individual spheres that compose each molecule. These interaction potentials cannot represent electrostatic potentials but can represent Lennard-Jones or harmonic repulsion type potentials.
Hybrid simulation
-
sp. Gillespie, PDE
-
-
-
Smoldyn allows particle and spatial Gillespie methods to be combined in a simulation. It can use an adjacent-space approach, in which adjacent regions are simulated with different methods [27]. It can also use an overlapping-space approach in which the same region of space is simulated with both methods, where some molecular species are simulated with one method other molecular species with the other method. The overlapping-space method has not been published yet. Smoldyn has also been incorporated into the VCell simulator, a simulator that was originally designed for deterministic spatial simulations using partial differential equations (PDEs). VCell's PDE solver works together with Smoldyn's particle-based solver to offer hybrid PDE and particle-based simulation [28].
Benchmark run time
67 s
22 s
13 days†
9.1 months†
13 minutes
† These run times are not directly comparable to the other times.
I ran identical simulations on several of the simulators to compare run times. All simulations used exactly the same hardware (early 2013 MacBook Pro). The same benchmark was used in ref. [5]. These simulations perform the simple Michaelis-Menten enzymatic reaction E+S <=> ES -> P using the following parameters: the association rate constant is kfwd = 5.88e6 M-1s-1 (0.01 μm3/s), the dissociation rate constant is kback = 1 s-1, and the product reaction rate constant is kprod = 1 s-1. All species were assigned a diffusion coefficient of 10 μm2s-1 and simulations ran for 10 s of simulated time. Systems were cubical with each side equal to 4.4964 μm for a total volume of 90.9 μm3 and simulations were started with 909 randomly placed E molecules (10 μm-3) and 9091 randomly placed S molecules (100 μm-3), which add to a total of 10,000 starting molecules. Where appropriate, time steps were 0.001 s, so simulators performed 10,000 iterations. I tried to optimize the simulator settings to yield the fastest times possible. Except as noted, all simulators returned essentially the same results, which agreed well with mass action calculations.
MCell was fastest with 11 partitions in each dimension. Input file: MCellBenchmark.txt (suffix should be changed to "mdl" for use).
Smoldyn was fastest with 15 partitions in each dimension. Input file: SmoldynBenchmark.txt.
eGFRD is not directly comparable to MCell, Smoldyn, and ReaDDy times because those simulators ignored excluded volume interactions whereas eGFRD accounted for them (using 1 nm molecule radii). Nevertheless, I simulated a model that was as similar as possible to the ones tested elsewhere and found that it took 18 minutes to complete 0.01 seconds of simulated time using eGFRD. From this, I computed that the total run would take about 13.0 days. I didn't check the simulation results for accuracy. Input file: eGFRDBenchmark.txt (suffix should be changed to "gfrd" for use). This difference in run times between eGFRD and other simulators was consistent with tests of bimolecular reactions (described in the reaction accuracy row of this table) and with results found when investigating macromolecular crowding with different simulators [16].
SpringSaLaD is even slower yet. It ran 0.1 ms of simulated time in 4 minutes, implying that it would have taken about 9.1 months to run 10 s of simulated time. Again, this is not directly comparable to results from the other simulators. In this case, SpringSaLaD used much smaller time steps than the other simulators (1 ns steps for spring dynamics and 10 ns for other dynamics, compared to 1 ms for the other simulators). Input file: SpringSaLaDBenchmark.txt.
ReaDDy worked well. This simulation ignored excluded volume interactions, making it faster than it would have been if it had accounted for these interactions, as it was primarily designed for. ReaDDy is clearly much slower than MCell or Smoldyn for this test, arising from the facts that it works at a higher level of detail and it was not optimized for this type of reaction network simulation. Input file: ReaDDyBenchmark.txt (suffix should be changed to "py" for use).
Distribution
executable
executable
self-compile
Java file
self-compile
Note that the software titles at the tops of the columns are links to the project websites. In most cases,
you can download from those web pages, or find instructions there.
MCell installs using pre-compiled binaries. The Mac download file is simply the executable binary, with no other installation required.
Smoldyn installs on Mac or Windows with pre-compiled binary files and an installer script. This is generally very easy. Smoldyn does not come with pre-compiled files for Linux, but the user needs to compile from the source using CMake there. Smoldyn has few dependencies, so compiling is generally quite straightforward.
eGFRD requires compiling using CMake. It depends upon the Gnu Scientific Library (GSL). On Macs, the basic simulation code builds well, but the eGFRD visualizer does not build due to calls to glut functions that are not supported by Macs.
ReaDDy requires local compilation. It installs relatively easily using the the miniconda package manager.
User interface
GUI, text
text
text
GUI
Python script
MCell is typically run with the CellBlender graphical user interface (GUI). It can also be run with text files, written in MCell's "model description language". MCell has extensive documentation.
Smoldyn input is always from text files. The input language is simple and straightforward. It is described thoroughly in the Smoldyn User's Manual.
eGFRD input is from text. The language is fairly self-explanatory, but has not been described in documentation yet.
SpringSaLaD input is from their own GUI. It outputs the simulation model to reasonably human-readable text files which are then run in the simulator.
ReaDDy is not a stand-alone program but a Python library. It is run by importing it into Python and then writing a script for the simulation.
Graphical output
excellent
good
partial support
partial support
good
MCell saves visualization data during the simulation, which can be viewed later but not during the simulation. MCell uses the CellBlender interface, which uses the free Blender package. The graphical quality can be excellent and has been used for making impressive movies.
Smoldyn typically presents a graphical display during the simulation. Graphics can be turned off if desired or can be displayed at three different quality levels, where low quality runs faster and high quality is slow but good for publications. Displayed images can be automatically stored to create still images or movies. Smoldyn can also store data for viewing afterwards using the VTK software.
eGFRD graphics can be visualized with gfrdVisualizer. It doesn't compile on a Mac because it uses glut library functions that aren't available in the Mac version of the glut library.
SpringSaLaD comes with an interactive three-dimensional viewer. However, it only works on Windows and Linux computers and not Macs [10].
ReaDDy output is available in the form of data, which can be plotted in real-time with other software, and a VMD compatible file that is generated after the simulation is over. (personal communication with Moritz Hoffmann).
Library interface
Python
C/C++
-
-
Python
MCell has an experimental Python interface that is still under development.
Smoldyn has a C/C++ language application programming interface (API) for the software [13]. This enables the Smoldyn physics engine to be included within other software. In particular, Smoldyn is included within the MOOSE neural simulation software.
ReaDDy is essentially nothing but a library API.
* The eGFRD algorithm is exact but the software doesn't simulate reaction rates correctly at present.
† These run times are not directly comparable to the other times.
To potential users:
Each of these programs is a high quality simulator that has been developed over many years. All of them are open-source and have non-restrictive licenses that allow for both commercial and non-commercial use. If one of them doesn't fit your needs, I suggest that you contact the appropriate developers to see if the software can be adapted in a way that does meet your needs. I strongly discourage writing new code until exhausting other options because doing so tends to consume multiple years of time and results in inferior software. Witness the long list of generally good but largely abandoned simulators: ChemCell, GMP, CDS, Meredys, SmartCell, GridCell, Cell++, SRSim, ...
To colleagues:
Please let me know if any table entries or annotations are incorrect.
Citation
This is an expanded version of a table that was published in the following article.
Steven S. Andrews, "Particle-based stochastic simulators" Encyclopedia of Computational Neuroscience, 2018. pdf
Acknowledgements
This comparison was made possible by assistance from: Jim Faeder and Jacob Czech (MCell), Marco Seynen and Pieter Rein ten Wolde (eGFRD), Paul Michalski and Les Loew (SpringSaLaD), and Frank Noe and Moritz Hoffman (ReaDDy).
References
- 1. R.A. Kerr, et al., Fast Monte Carlo simulation methods for biological reaction-diffusion systems in solution and on surfaces. SIAM J. Sci. Comput. 30, 3126-3149 (2008).
- 2. T.M. Bartol, et al., Computational reconstitution of spine calcium transients from individual proteins. Frontiers in Synaptic Neuroscience 7, 17 (2015).
- 3. J.P. Kinney, et al., Extracellular sheets and tunnels modulate glutamate diffusion in hippocampal neuropil. J. Comparative Neurology 521, 448-464 (2013).
- 4. J.S. Coggan, et al., Evidence for ectopic neurotransmission at a neuronal synapse. Science 309, 446-451 (2005).
- 5. S.S. Andrews, N.J. Addy, R. Brent, and A.P. Arkin, Detailed simulation of cell biology with Smoldyn 2.1. PLoS Comp. Biol. 6, e1000705 (2010).
- 6. S.S. Andrews, Spatial and stochastic cellular modeling with the Smoldyn simulator. Methods in Molecular Biology 804, 519-542 (2012).
- 7. J.S. van Zon and P.R. ten Wolde, Green's function reaction dynamics: A particle-based approach for simulating biochemical networks in time and space. J. Chem. Phys. 123, 234910 (2005).
- 8. J.S. van Zon and P.R. ten Wolde, Simulating biochemical networks at the particle level in time and space: Green's function reaction dynamics. Phys. Rev. Lett. 94, 128103 (2005).
- 9. K. Takahashi, S. Tanase-Nicola, and P.R. ten Wolde, Spatio-temporal correlations can drastically change the response of a MAPK pathway. Proc. Natl. Acad. Sci. USA 107, 2473-2478 (2010).
- 10. P.J. Michalski and L.M. Loew, SpringSaLaD: a spatial, particle-based biochemical simulation platform with excluded volume. Biophys J. 110, 523-529 (2016).
- 11. J. Schöneberg and F. Noé, ReaDDy - A software for particle-based reaction-diffusion dynamics in crowded cellular environments. PLoS ONE 8, e74261 (2013).
- 12. S.S. Andrews and D. Bray, Stochastic simulation of chemical reactions with spatial resolution and single molecule detail. Phys. Biol. 1, 137-151 (2004).
- 13. S.S. Andrews, Smoldyn: particle-based simulation with rule-based modeling, improved molecular interaction and a library interface. Bioinformatics 33, 710-717 (2017).
- 14. T.R. Sokolowski, et al., eGFRD in all dimensions. ArXiv 1708, 09364v1 (2017).
- 15. T. Radó, Über den begriff der Riemannschen fläche. Acta Litt. Sci. Szeged 2, 101-121 (1925).
- 16. S.S. Andrews, et al., Simulating macromolecular crowding with particle and lattice-based methods, in Dagstuhl Reports: Multiscale Spatial Computational Systems Biology (Dagstuhl Seminar 14481), D. Gilbert, et al., Editors. p. 170-187 (2015).
- 17. M.I. Stefan, T.M. Bartol, T.J. Sejnowski, and M.B. Kennedy, Multi-state modeling of biomolecules. PLoS Comp. Biol. 10, e1003844 (2014).
- 18. S.S. Andrews, Rule-based modeling using wildcards in the Smoldyn simulator. Methods in Molecular Biology In press (2018).
- 19. T.A.J. Duke, N. LeNovère, and D. Bray, Conformational spread in a ring of proteins: a stochastic approach to allostery. J. Mol. Biol. 308, 541-553 (2001).
- 20. R. Erban and S.J. Chapman, Stochastic modelling of reaction-diffusion processes: algorithms for bimolecular reactions. Phys. Biol. 6, 46001 (2009).
- 21. T.R. Sokolowski and P.R. ten Wolde, Spatial-stochastic simulation of reaction-diffusion systems. ArXiv 1705, 08669 (2017).
- 22. F.C. Collins and G.E. Kimball, Diffusion-controlled reaction rates. J. Colloid Sci. 4, 425-437 (1949).
- 23. S.S. Andrews, Serial rebinding of ligands to clustered receptors as exemplified by bacterial chemotaxis. Phys. Biol. 2, 111-122 (2005).
- 24. S.A. Rice, Diffusion Limited Reactions. Vol. 25, Elsevier: Amsterdam (1985).
- 25. R. Erban and S.J. Chapman, Reactive boundary conditions for stochastic simulations of reaction-diffusion processes. Phys. Biol. 4, 16-28 (2007).
- 26. S.S. Andrews, Accurate particle-based simulation of adsorption, desorption, and partial transmission. Phys. Biol. 6, 46015 (2009).
- 27. M. Robinson, S.S. Andrews, and R. Erban, Multiscale reaction-diffusion simulations with Smoldyn. Bioinformatics 31, 2406-2408 (2015).
- 28. J.C. Schaff, et al., Numerical approach to spatial deterministic-stochastic models arising in cell biology. PLoS Comp. Biol. 12:e1005236 (2016)