
What Smoldyn isSmoldyn is a computer program for cell-scale biochemical simulations. It simulates each molecule of interest individually to capture natural stochasticity and to yield nanometer-scale spatial resolution. It treats other molecules implicitly, enabling it to simulate hundreds of thousands of molecules over several minutes of real time. Simulated molecules diffuse, react, are confined by surfaces, and bind to membranes much as they would in a real biological system. Smoldyn is easy to use and easy to install. It is more accurate and faster than other particle-based simulators. Smoldyn's features include: simulations in 1, 2, or 3 dimensions, a "virtual experimenter" who can manipulate or measure the simulated system, molecules with excluded volume, rule-based modeling, and Python bindings. |
NewsSmoldyn 2.74 released Jan. 22, 2025
|
Research Highlight
|
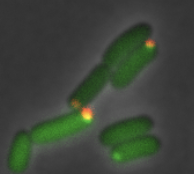
Phospho-signaling couples polar asymmetry and proteolysis within a membraneless microdomain in Caulobacter crescentusAhmed, Y.M. and G.R. Bowman Nature Comm. 15:9282, 2024Many bacterial species, including the C. crescentus bacterium investigated here, exhibit asymmetric cell division, leading to the question of how the cell directs specific regulatory proteins to the correct cell poles. Curiously, both poles contain a membraneless phase-separated microdomain, established by the polar assembly hub PopZ, through most of the cell cycle, yet many of the proteins that depend on it exhibit unipolar and transient localization. Based on many experimental observations, the authors hypothesized that the activity of the kinase/phosphatase protein CckA acts as a switch that regulates PopZ's interaction several other proteins, and that this switch is controlled by a protein that localizes to only one of the two cell poles. This hypothesis was confirmed by experiement and particle-based modeling. |
Smoldyn is written and maintained by Steve Andrews. Development has been supported by the National Institutes of Health, the U.S. Department of Energy, the National Science Foundation, and the MITRE Corporation, albeit never by funding that was dedicated specifically for this purpose.


![]()
![]()